Epigenetic Clocks Predict Disease 30 Years Early

Epigenetic Clocks Predict Disease 30 Years Early
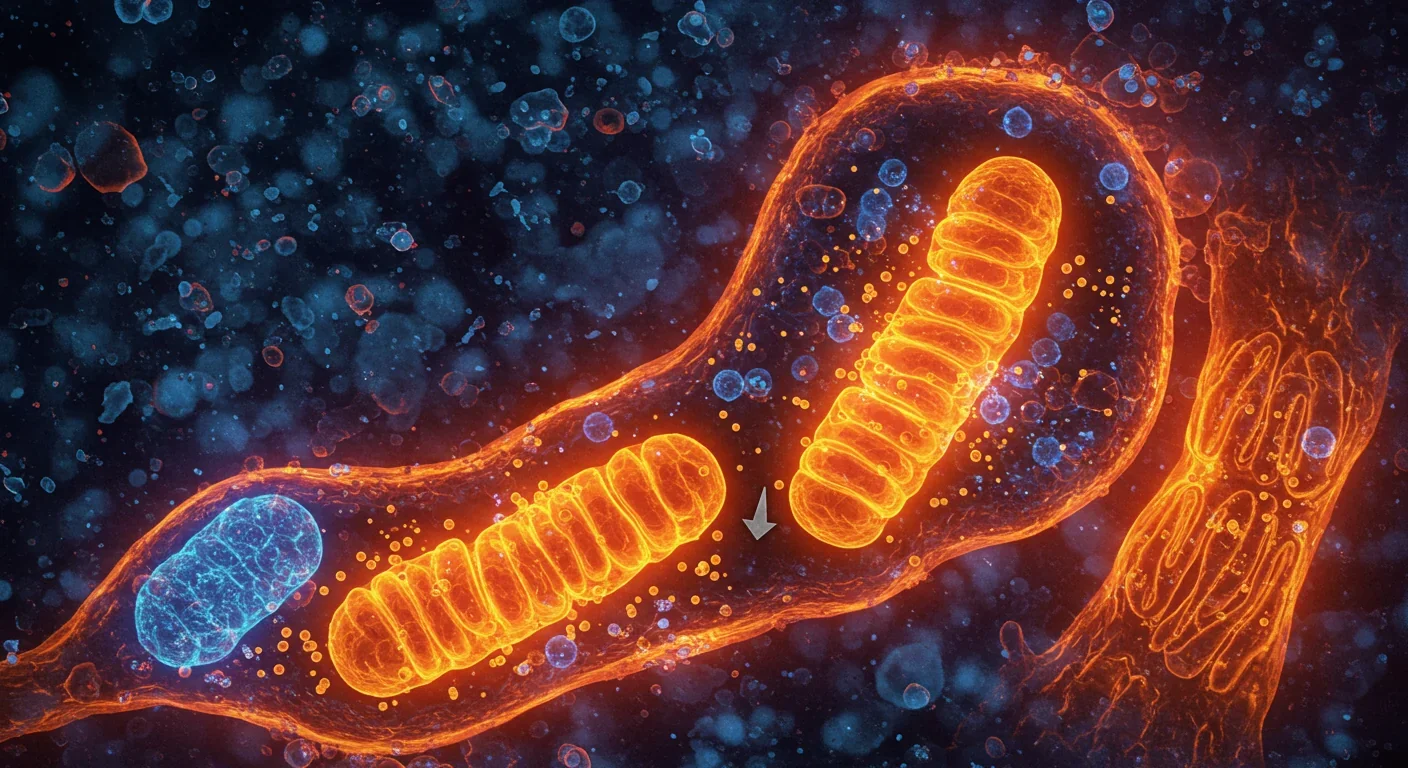
Mitophagy: The Cellular Cleanup That Prevents Aging

Interoception Training: Stop Anxiety & Chronic Pain Naturally

Parasitic Castration: Nature's Reproductive Hijacking

Interoception: The Hidden Sense Controlling Your Emotions

Base Editing: Safer Gene Therapy Rewriting DNA Letter by Letter
TL;DR: Senescent "zombie" cells—cells that stop dividing but refuse to die—accumulate with age and secrete inflammatory molecules that damage neighboring tissues, driving chronic diseases like arthritis, Alzheimer's, and heart disease. Comprising only 2–3% of tissue mass, these cells act as master regulators of aging. New senolytic drugs that selectively eliminate them have extended lifespan by 20–30% in mice and are now in human trials, with early successes restoring vision in diabetic patients. Lifestyle factors—exercise, Mediterranean diet, sleep, fasting—also slow senescent cell buildup. The future promises precision therapies guided by MRI imaging and aging clocks, potentially compressing decades of frailty into a brief twilight and rewriting the rules of human healthspan.

By 2030, scientists predict that one in five people worldwide will be over 60, and the majority will carry billions of "zombie cells" in their bodies—cells that refuse to die, accumulate with age, and silently fuel chronic diseases from arthritis to Alzheimer's. These senescent cells, as researchers call them, have stopped dividing but won't trigger their own death, lingering instead in tissues where they secrete a toxic cocktail of inflammatory molecules. For decades, biologists dismissed them as passive bystanders of aging. Now, mounting evidence reveals they are architects of our decline, and a new class of drugs designed to eliminate them could rewrite the rules of human healthspan.
In 1961, anatomist Leonard Hayflick discovered that normal human cells divide only 40 to 60 times before entering a permanent growth arrest—a phenomenon now known as the Hayflick limit. For years, this was interpreted as a simple biological clock: cells run out of steam, stop dividing, and fade into the background. But recent research paints a far more sinister picture. Senescent cells don't fade quietly. Instead, they become metabolically hyperactive, secreting a potent mix of inflammatory cytokines (IL-6, IL-8), growth factors (IGFBPs), and tissue-remodeling enzymes collectively termed the senescence-associated secretory phenotype, or SASP.
This secretory profile doesn't just mark cellular retirement—it actively damages neighboring healthy cells, spreads senescence through paracrine signaling, and degrades the extracellular matrix that holds tissues together. In 2015, researchers at the Mayo Clinic and Scripps Research demonstrated that genetically clearing senescent cells from aged mice extended their median lifespan by 20–30% and reversed age-related functional decline. Mice treated with senolytic drugs—compounds that selectively kill zombie cells—showed improved heart function, stronger muscles, and reduced cancer risk. The implications were staggering: senescent cells, though comprising only 2–3% of tissue mass, could be a master regulator of aging itself.
Today, the race is on. Over a dozen senolytic drugs are in human clinical trials, from repurposed cancer therapeutics like dasatinib combined with the plant flavonoid quercetin, to novel compounds such as UBX1325, a BCL-xL inhibitor that recently restored vision in patients with diabetic macular edema. Biological aging, once considered irreversible, is now a targetable disease state.
The concept of cellular senescence has undergone three major paradigm shifts. In the 1960s, Hayflick's work established replicative senescence as a tumor-suppressive mechanism: cells with critically short telomeres—protective DNA caps at chromosome ends—stop dividing to prevent malignant transformation. This was seen as a beneficial brake on cancer. By the 1990s, researchers recognized that senescence could also be triggered prematurely by DNA damage, oxidative stress, or oncogene activation, independent of telomere length. Yet senescent cells were still viewed as inert, post-mitotic husks.
The third shift began in the 2000s with the discovery of SASP. Scientists realized that senescent cells are anything but silent. They continuously pump out pro-inflammatory signals that recruit immune cells, alter tissue architecture, and induce senescence in bystander cells—a phenomenon called secondary or paracrine senescence. This feedback loop creates a spreading wave of dysfunction. For instance, the protein IGFBP7, a key SASP component, binds insulin and activin receptors on neighboring mesenchymal stem cells, pushing them into senescence within 48 hours. Neutralizing IGFBP7 with antibodies reduces secondary senescence from 35% to 10%, proving that SASP factors themselves—not just the senescent cells—drive tissue aging.
Historically, aging research borrowed lessons from infectious disease and cancer: identify a pathogen or mutation, then target it. Senescence offered a new model—aging as an accumulation of dysfunctional cells that could be selectively removed. This paralleled earlier breakthroughs in telomere biology (Elizabeth Blackburn and Carol Greider's Nobel Prize-winning discovery of telomerase in 1984) and the realization that chronic inflammation—"inflammaging"—underpins most age-related diseases. Together, these insights laid the groundwork for senotherapeutics: drugs that either eliminate senescent cells (senolytics) or dampen their inflammatory output (senomorphics).
Senescence isn't a single event but a complex program triggered by diverse stressors. The primary molecular pathways converge on two tumor-suppressor axes: p53-p21 and p16INK4a-Rb. When DNA damage, telomere shortening, or oncogenic signals activate the kinase ATM, it phosphorylates the protein p53, which in turn upregulates p21. This cyclin-dependent kinase inhibitor halts cell cycle progression, preventing damaged cells from replicating. Simultaneously, the cell accumulates p16INK4a, which locks the retinoblastoma (Rb) protein in an active, growth-suppressive state. Once both pathways engage, the cell enters irreversible arrest.
But why do senescent cells evade apoptosis—the programmed cell death that normally eliminates damaged cells? The answer lies in anti-apoptotic proteins. Senescent cells upregulate BCL-2, BCL-xL, and BCL-w, members of a protein family that blocks mitochondrial outer membrane permeabilization, the key step in apoptosis. These proteins act like molecular shields, allowing zombie cells to survive despite catastrophic internal damage. This survival mechanism is both their Achilles' heel and the basis for senolytic therapy: drugs that inhibit BCL family proteins selectively kill senescent cells while sparing healthy ones.
Telomere shortening is the best-studied trigger. Telomeres shorten by 50–100 base pairs with each cell division. Once they reach a critical length—typically 4,000–6,000 base pairs in humans—the DNA damage response is activated, mistaking the short telomere for a double-strand break. The enzyme telomerase, a reverse transcriptase that adds telomeric repeats, is active in germ cells and stem cells but repressed in most somatic tissues. This asymmetry explains why tissues with high turnover—bone marrow, gut epithelium, skin—accumulate senescent cells faster than post-mitotic tissues like neurons (which face different senescence triggers, such as protein aggregation and mitochondrial dysfunction).
Oxidative stress amplifies telomere attrition. Telomeric DNA is rich in guanine residues, which are highly vulnerable to reactive oxygen species (ROS). A single oxidative hit can delete 200–500 base pairs, effectively accelerating the aging clock. Chronic inflammation, a hallmark of Western dietary patterns high in processed foods and low in antioxidants, floods tissues with ROS, shortening telomeres and driving premature senescence. In contrast, adherence to a Mediterranean diet—rich in polyphenols, omega-3 fatty acids, and fiber—has been shown to increase telomerase activity and extend leukocyte telomere length by the equivalent of 4.5 biological years.
Oncogene-induced senescence (OIS) represents another pathway. When cells experience aberrant activation of growth signals—such as mutant RAS or overexpressed MYC—they initially proliferate rapidly, then abruptly arrest due to replication stress and DNA damage. OIS serves as a fail-safe against cancer, but the senescent cells that result contribute to tissue aging and, paradoxically, can later promote tumor progression through SASP-mediated remodeling of the tumor microenvironment.
The senescence-associated secretory phenotype is the primary mechanism by which a few senescent cells wreak havoc on entire tissues. SASP factors include:
Inflammatory cytokines: IL-6, IL-8, IL-1β, TNF-α
Chemokines: MCP-1, CXCL-1
Growth factors: IGFBPs, VEGF, HGF
Matrix metalloproteinases: MMP-3, MMP-9, which degrade collagen and elastin
Extracellular vesicles: carrying microRNAs, DNA fragments, and proteins like CAP1
SASP composition varies by cell type and inducing stressor. For example, senescent fibroblasts secrete high levels of matrix-remodeling enzymes, while senescent vascular smooth muscle cells produce factors that stiffen arteries. This heterogeneity complicates therapeutic targeting but also offers opportunities for precision interventions.
Two transcription factors—NF-κB and C/EBPβ—orchestrate SASP gene expression. Persistent DNA damage activates ATM, which phosphorylates p53 and upregulates p21. p21 inhibits CDK2, preventing phosphorylation and inactivation of the E3 ubiquitin ligase complex Cdh1:APC/C. Active APC/C then targets the histone methyltransferases EHMT1 and EHMT2 for degradation. These enzymes normally repress IL6 and IL8 gene promoters by maintaining repressive histone methylation marks. Their loss opens the floodgates: IL-6 and IL-8 transcription surges, IL-6 signaling stimulates C/EBPβ transcription, and C/EBPβ further amplifies IL-6 and IL-8 expression, creating a positive feedback loop that locks in the inflammatory phenotype.
SASP also exerts autocrine effects, reinforcing the senescent state within the producing cell, and paracrine effects, inducing senescence in neighboring healthy cells. IGFBP7, for instance, is secreted by senescent mesenchymal stem cells and binds to insulin, IGF-II, and activin A in the extracellular space, modulating their signaling. IGFBP7 blocks insulin's anti-senescence signals, promotes IGF-II signaling via IGF2R (which lacks anti-senescence activity), and engages activin receptors ACVR1 and ACVR1B, activating SMAD2/3 and SMAD1/5 pathways that drive senescence in recipient cells. This cascade exemplifies how a single SASP factor can reprogram the metabolic and signaling environment to propagate cellular aging.

Extracellular vesicles (EVs) add another dimension. Senescent cells release EVs enriched in pro-inflammatory cargo, including the actin-binding protein CAP1. When EVs from aged, atherosclerotic mice are injected into young wild-type mice, they induce endothelial senescence, β-galactosidase staining, and even plaque formation in arteries—despite a normal diet. This suggests that senescence can spread systemically via circulating EVs, explaining how localized senescent cell burden in one organ (e.g., adipose tissue) can accelerate aging in distant tissues (e.g., brain, heart).
Cellular senescence is causally implicated in nearly every major age-related disease. Here's how zombie cells drive specific pathologies:
Cardiovascular Disease: Senescent vascular smooth muscle cells and endothelial cells accumulate in arteries with age. They secrete matrix metalloproteinases that degrade elastin and collagen, increasing arterial stiffness—a key predictor of hypertension and stroke. Senescent endothelial cells also upregulate adhesion molecules that promote leukocyte infiltration and plaque formation. Telomerase activity, paradoxically, has been observed in atherosclerotic plaques, where it may support the survival of dysfunctional cells rather than healthy regeneration. In mouse models, clearing p16-positive senescent cells with genetic ablation or senolytics reduces plaque burden and restores vascular compliance.
Osteoarthritis: Cartilage is an essentially post-mitotic tissue, but chondrocytes (cartilage cells) can still enter senescence due to mechanical stress and oxidative damage. Senescent chondrocytes secrete IL-1, IL-6, and MMPs that degrade cartilage matrix, perpetuating joint inflammation and pain. In pig models, MRI contrast agents that selectively illuminate senescent cells (by targeting the enzyme β-galactosidase, which is highly expressed in zombie cells) have successfully visualized senescent cell burden in arthritic joints. This imaging breakthrough could enable rapid assessment of senolytic drug efficacy in clinical trials, replacing long-term functional endpoints with near-real-time biomarkers.
Alzheimer's Disease and Neurodegeneration: Microglia—the brain's resident immune cells—can become senescent, adopting a pro-inflammatory phenotype that impairs their ability to clear amyloid-β plaques and hyperphosphorylated tau tangles. In the PS19 mouse model of tauopathy, genetic elimination of p16-positive senescent microglia reduces tau aggregation, neurofibrillary tangle load, and cognitive decline. Senescent astrocytes, which support neuronal metabolism, also secrete SASP factors that disrupt the blood-brain barrier and promote neuronal death. The interplay between senescent glia, misfolded proteins, and neuroinflammation forms a vicious cycle that accelerates neurodegenerative disease.
Type 2 Diabetes: Senescent cells accumulate in pancreatic islets, adipose tissue, and liver with obesity and aging. In pancreatic β-cells, senescence impairs insulin secretion. In adipose tissue, senescent adipocytes secrete inflammatory cytokines that promote insulin resistance in muscle and liver. Metformin, a first-line diabetes drug, exerts senomorphic effects by activating AMPK and inhibiting mTOR, pathways that suppress SASP production and enhance autophagy—the cellular recycling process that clears damaged organelles. Although metformin does not kill senescent cells, it reduces their inflammatory output and has been associated with increased healthspan in diabetic patients.
Cancer: Senescence has a dual role in cancer. Initially, oncogene-induced senescence acts as a tumor suppressor, halting the proliferation of pre-malignant cells. However, the SASP from these senescent cells can later promote tumor progression by remodeling the extracellular matrix, recruiting immunosuppressive myeloid cells, and providing growth factors that support tumor angiogenesis and metastasis. Senescent stromal cells in the tumor microenvironment upregulate PD-L1, creating a T-cell-excluded niche that blunts anti-tumor immunity. This paradox has spurred interest in combining senolytics with cancer immunotherapy: clearing senescent cells may restore immune surveillance and enhance response to checkpoint inhibitors.
Eye Diseases: Lens epithelial cells undergo senescence with age, contributing to cataract formation. In diabetic macular edema (DME) and age-related macular degeneration, senescent retinal cells secrete VEGF and inflammatory mediators that drive vascular leakage and vision loss. UBX1325, a BCL-xL inhibitor, delivered via intravitreal injection, selectively eliminates senescent cells in the diabetic retina. In the Phase 2 BEHOLD study, a single 10-microgram dose of UBX1325 improved mean best-corrected visual acuity by over 15 ETDRS letters at 48 weeks compared to sham treatment—a sustained, clinically meaningful benefit from eliminating zombie cells.
Senolytic drug development has accelerated dramatically since 2015. The field now includes repurposed compounds, natural products, and novel targeted agents:
Dasatinib + Quercetin (D+Q): Dasatinib, a tyrosine kinase inhibitor approved for chronic myeloid leukemia, targets senescent cell anti-apoptotic pathways, including BCL-2 and PI3K/AKT. Quercetin, a flavonoid found in onions and apples, inhibits BCL-xL and other survival factors. Together, they selectively induce apoptosis in senescent cells. In human pilot trials, D+Q administered intermittently (two consecutive days, then off for weeks) reduced circulating SASP markers and improved physical function in patients with idiopathic pulmonary fibrosis. A current trial at Mayo Clinic and Cedars-Sinai is testing D+Q in patients with schizophrenia and treatment-resistant depression, conditions associated with accelerated biological aging and elevated senescence burden.
Fisetin: This plant flavonoid, abundant in strawberries and apples, is one of the most potent natural senolytics. Fisetin reduces senescent cell markers (p16, p21, β-galactosidase) and lowers inflammatory cytokines IL-6 and TNF-α in mouse arteries. A 2024 study in Aging Cell found that intermittent fisetin dosing (one week on, two weeks off) cleared senescent cells and improved vascular function. Human trials use 20 mg/kg daily for two consecutive days per month. Fisetin also acts as an antioxidant and sirtuin activator, making it a bridge between dietary intervention and targeted senolytic therapy. However, bioavailability is low unless co-administered with dietary fats like olive or coconut oil.
Navitoclax (ABT-263): A potent BCL-2/BCL-xL/BCL-w inhibitor developed for cancer, navitoclax clears senescent cells in preclinical models and reverses immunosuppression in the tumor microenvironment, restoring CD8+ T-cell proliferation. Its dose-limiting toxicity—thrombocytopenia due to on-target platelet inhibition—has limited systemic use, but intermittent dosing or local delivery (e.g., intra-articular injection for osteoarthritis) may mitigate this.
UBX1325: Unity Biotechnology's BCL-xL inhibitor is designed for local administration to avoid systemic toxicity. Delivered by intravitreal injection, UBX1325 selectively eliminates senescent cells in the retina. In DME patients, visual acuity gains persisted through 48 weeks after a single dose, demonstrating that senolytic-induced tissue remodeling can produce durable functional improvements. Unity is also developing UBX1967 for other ophthalmologic indications. These successes highlight the feasibility of tissue-specific senolytic therapy.
p53 Activators: Rather than killing senescent cells, these compounds restore p53 function, which suppresses SASP by blocking the formation of cytoplasmic chromatin fragments (CCF)—bits of damaged DNA that trigger inflammatory signaling via the cGAS-STING pathway. In aged mice, a p53-activating drug reversed the SASP transcriptional signature without reducing senescent cell number, suggesting that SASP suppression alone can mitigate inflammation and tissue dysfunction. This senomorphic approach may be safer for long-term use than ablative senolytics.
Senolytics in Clinical Trials: Over 20 senolytic trials are registered on ClinicalTrials.gov, targeting conditions including osteoarthritis, chronic kidney disease, Alzheimer's disease, frailty, and COVID-19 long-haul syndrome (where viral infection induces widespread senescence). Most trials use intermittent "hit-and-run" dosing to minimize toxicity while achieving senescent cell clearance.
Beyond pharmacological interventions, several lifestyle factors modulate senescent cell accumulation:
Exercise: Physical activity activates natural killer (NK) cells and CD8+ T cells, which recognize and eliminate senescent cells via NKG2D receptor engagement with stress ligands MICA and ULBP2 on zombie cells. A 12-week aerobic exercise program in older adults reduced p16INK4a expression in muscle biopsies. Exercise also triggers autophagy and mitophagy—processes that clear damaged mitochondria, reducing ROS production and senescence triggers.
Caloric Restriction and Intermittent Fasting: These interventions activate AMPK and sirtuins, suppress mTOR, and enhance autophagy, all of which reduce senescent cell burden. In mice, intermittent fasting combined with 20% caloric restriction accelerates senescent cell clearance and improves markers of tissue function. Fasting induces a metabolic switch to ketone body utilization, producing β-hydroxybutyrate, which inhibits NLRP3 inflammasome activation—a key driver of SASP.
Mediterranean Diet: High in polyphenols (from olive oil, red wine, berries), omega-3 fatty acids (from fish), and fiber, this dietary pattern increases telomerase activity, reduces oxidative stress, and lowers systemic inflammation. A cohort study found that adherence to the Mediterranean diet was associated with leukocyte telomere lengths 1.5 years biologically younger than age-matched controls on a Western diet. Specific nutrients—resveratrol, curcumin, quercetin—have senolytic or senomorphic properties.
Sleep: Chronic sleep deprivation elevates cortisol and inflammatory cytokines, accelerates telomere shortening, and impairs autophagy. Seven to nine hours of quality sleep per night supports circadian-regulated DNA repair and immune clearance of senescent cells.
Stress Management: Psychological stress increases telomerase activity acutely (an adaptive response) but chronic stress shortens telomeres and induces senescence in immune cells. Mindfulness-based stress reduction and cognitive-behavioral therapy have been shown to preserve telomere length and reduce inflammatory biomarkers in clinical trials.
NAD+ Boosting: Nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) are NAD+ precursors that activate sirtuins and PARPs, enhancing DNA repair and mitochondrial function. In Werner syndrome fibroblasts (a model of premature aging), NR supplementation reduced senescence markers and restored metabolic gene expression. However, NR did not rescue proliferative capacity in immortalized cells, indicating that NAD+ repletion addresses metabolic dysfunction but not the fundamental proliferative arrest of senescence.
Despite their promise, senolytics raise important safety and ethical questions:
Beneficial Senescence: Not all senescent cells are harmful. During wound healing, a transient wave of senescent fibroblasts secrete growth factors and matrix-remodeling enzymes that facilitate tissue repair. In embryonic development, programmed senescence helps sculpt tissues. Chronic, indiscriminate senolytic therapy could impair these beneficial processes. Current evidence suggests that the senescent cells that accumulate with aging are distinct from those in acute repair, but more research is needed to identify context-specific markers.
Immune Suppression: Senescent cells secrete chemokines that recruit immune cells. While this is pathological in chronic inflammation, it may also support immune surveillance against pathogens and tumors. Clearing senescent immune cells could impair immune function. Conversely, eliminating senescent stromal cells that produce immunosuppressive signals (e.g., PD-L1) might enhance immunity.
Off-Target Toxicity: BCL-2/BCL-xL inhibitors affect platelets, which rely on BCL-xL for survival, causing thrombocytopenia. Local delivery and intermittent dosing mitigate this, but systemic senolytic therapy requires careful monitoring. The therapeutic window—dose sufficient to clear senescent cells without harming healthy ones—varies by drug and tissue.
Rebound Senescence: If senolytics trigger compensatory proliferation in stem cell compartments, and those cells encounter unresolved DNA damage, they may rapidly re-enter senescence. Combining senolytics with interventions that enhance DNA repair or reduce oxidative stress (e.g., NAD+ precursors, antioxidants) may prevent rebound.
Equity and Access: Senolytic therapies, if proven effective, could exacerbate health disparities. Expensive drugs and precision diagnostics (e.g., MRI-based senescence imaging) may be accessible only to wealthy populations, widening the longevity gap. Affordable, over-the-counter senolytic supplements (e.g., fisetin, quercetin) are unregulated, with variable quality and unproven dosing, posing risks to consumers seeking DIY anti-aging interventions.
Lifespan Extension: If senolytics significantly extend human healthspan, societal structures—retirement, healthcare systems, workforce dynamics—will need to adapt. Ethical frameworks must address questions of resource allocation, intergenerational equity, and the desirability of radical life extension.
The next decade will see rapid advances in senolytic science:
Non-Invasive Imaging: MRI contrast agents that selectively illuminate senescent cells (via β-galactosidase or other markers) are in development. These will enable clinicians to quantify senescent cell burden in specific tissues, stratify patients by senescence load, and monitor drug efficacy in real time—replacing months-long functional endpoints with weeks-long imaging readouts.
Aging Clocks: DNA methylation-based aging clocks (e.g., Horvath, GrimAge) can measure biological age and predict mortality risk. Integrating aging clocks with senolytic trials will allow researchers to assess whether clearing senescent cells "rewinds" biological age. Preliminary data suggest senolytic therapy reduces epigenetic age by 1–2 years within months.
SASP Biomarkers: Blood-based assays measuring circulating SASP factors (IL-6, MMP-3, IGFBP7) or senescent-cell-derived EVs could serve as liquid biopsies for systemic senescence burden. These biomarkers would enable early detection of aging acceleration and personalized dosing of senolytics.
Combination Therapies: Senolytics may synergize with metformin (which suppresses SASP via AMPK/mTOR modulation), NAD+ boosters (which enhance mitochondrial function and DNA repair), and immune checkpoint inhibitors (which restore immune clearance of senescent cells). A "senolytic cocktail" tailored to individual senescence profiles—measured by imaging, biomarkers, and aging clocks—could optimize outcomes.
Gene Therapy: CRISPR/Cas9 editing to upregulate p53, suppress SASP transcription factors (NF-κB, C/EBPβ), or activate pro-apoptotic pathways selectively in senescent cells is in preclinical development. Alternatively, gene therapy to restore telomerase activity in specific tissues (without increasing cancer risk) could prevent replicative senescence.
Gut Microbiome Modulation: Emerging evidence links gut dysbiosis to systemic inflammation and senescence. Transferring fecal microbiota from young to old mice improves healthspan and reduces senescence markers. Probiotic or prebiotic interventions that reshape the microbiome may complement senolytics by reducing inflammation at its source.
For the first time in history, we possess the tools to target a fundamental mechanism of aging: the accumulation of senescent cells. What began as a curiosity—why do cells stop dividing?—has blossomed into a therapeutic revolution. Zombie cells, once thought to be inert debris, are now recognized as active agents of tissue destruction, spreading inflammation and dysfunction throughout the body. Drugs that selectively eliminate them are already restoring vision in diabetic patients, and trials are underway in diseases from Alzheimer's to arthritis.
The promise is profound: not merely adding years to life, but adding life to years—compressing morbidity into a brief twilight rather than enduring decades of frailty. A world where 80-year-olds hike mountains, where Alzheimer's rates plummet, where heart attacks become rare, is within reach if senolytics fulfill their potential.
Yet the science is young. We don't yet know the long-term effects of chronic senescent cell clearance, the optimal dosing schedules, or how to distinguish harmful zombie cells from beneficial ones. We must proceed with rigor, balancing the urgency of aging populations with the caution demanded by any intervention that alters fundamental biology.
As you read this, billions of senescent cells are secreting their toxic cargo into your tissues. Some are remnants of infections you fought decades ago. Others arose from last night's poor sleep or this morning's stressful commute. But unlike previous generations, you live in an era where these cells are no longer invisible and untouchable. Exercise, diet, sleep, and stress management can slow their accumulation. In the coming years, precision senolytics—guided by imaging and biomarkers—may selectively eliminate them, rewinding your biological clock.
The zombie apocalypse is already underway inside your body. The question is: will you be among the first to fight back?

Recent breakthroughs in fusion technology—including 351,000-gauss magnetic fields, AI-driven plasma diagnostics, and net energy gain at the National Ignition Facility—are transforming fusion propulsion from science fiction to engineering frontier. Scientists now have a realistic pathway to accelerate spacecraft to 10% of light speed, enabling a 43-year journey to Alpha Centauri. While challenges remain in miniaturization, neutron management, and sustained operation, the physics barriers have ...
Epigenetic clocks measure DNA methylation patterns to calculate biological age, which predicts disease risk up to 30 years before symptoms appear. Landmark studies show that accelerated epigenetic aging forecasts cardiovascular disease, diabetes, and neurodegeneration with remarkable accuracy. Lifestyle interventions—Mediterranean diet, structured exercise, quality sleep, stress management—can measurably reverse biological aging, reducing epigenetic age by 1-2 years within months. Commercial ...

Data centers consumed 415 terawatt-hours of electricity in 2024 and will nearly double that by 2030, driven by AI's insatiable energy appetite. Despite tech giants' renewable pledges, actual emissions are up to 662% higher than reported due to accounting loopholes. A digital pollution tax—similar to Europe's carbon border tariff—could finally force the industry to invest in efficiency technologies like liquid cooling, waste heat recovery, and time-matched renewable power, transforming volunta...

Humans are hardwired to see invisible agents—gods, ghosts, conspiracies—thanks to the Hyperactive Agency Detection Device (HADD), an evolutionary survival mechanism that favored false alarms over fatal misses. This cognitive bias, rooted in brain regions like the temporoparietal junction and medial prefrontal cortex, generates religious beliefs, animistic worldviews, and conspiracy theories across all cultures. Understanding HADD doesn't eliminate belief, but it helps us recognize when our pa...

The bombardier beetle has perfected a chemical defense system that human engineers are still trying to replicate: a two-chamber micro-combustion engine that mixes hydroquinone and hydrogen peroxide to create explosive 100°C sprays at up to 500 pulses per second, aimed with 270-degree precision. This tiny insect's biochemical marvel is inspiring revolutionary technologies in aerospace propulsion, pharmaceutical delivery, and fire suppression. By 2030, beetle-inspired systems could position sat...

The U.S. faces a catastrophic care worker shortage driven by poverty-level wages, overwhelming burnout, and systemic undervaluation. With 99% of nursing homes hiring and 9.7 million openings projected by 2034, the crisis threatens patient safety, family stability, and economic productivity. Evidence-based solutions—wage reforms, streamlined training, technology integration, and policy enforcement—exist and work, but require sustained political will and cultural recognition that caregiving is ...

Every major AI model was trained on copyrighted text scraped without permission, triggering billion-dollar lawsuits and forcing a reckoning between innovation and creator rights. The future depends on finding balance between transformative AI development and fair compensation for the people whose work fuels it.

Loading featured articles...